Introduction¶
Although intially designed for selecting protein markers, ProMS can be used for selecting discriminative features in any target view with or without the information from auxiliary views. Target variable can be binary (classification), continuous (regression) or censored survival data (survival analysis). In the following, we will use protein marker selection for classification as an example to describe the algorithms.
We provide two methods for selecting protein markers.
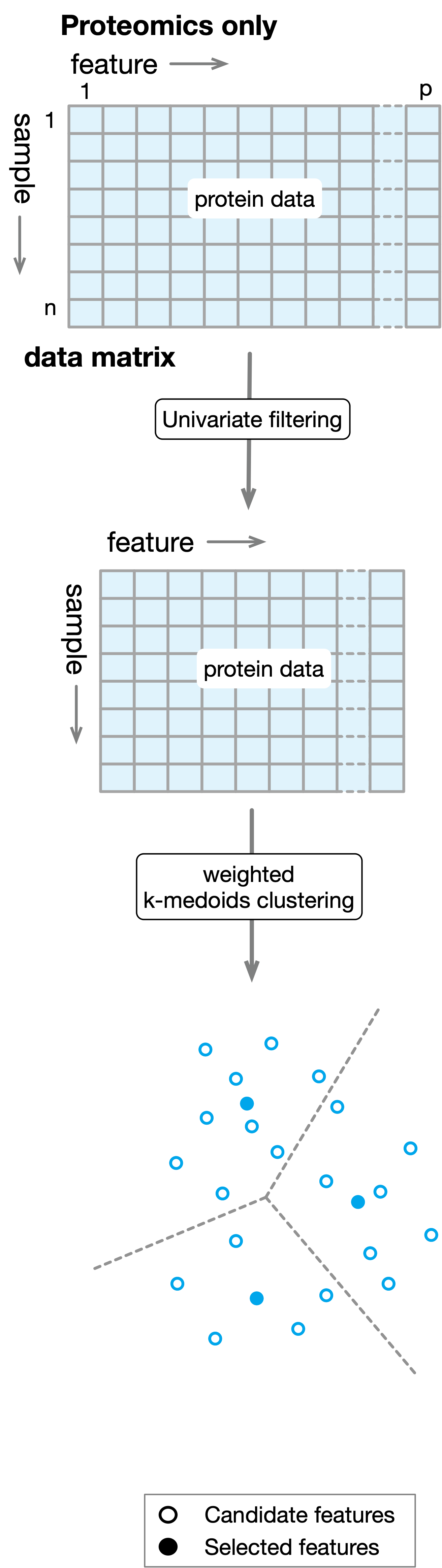
ProMS: Protein marker selection with proteomics data alone¶
The algorithm ProMS works as follows. As a first step to remove uninformative features, ProMS examines each feature individually to determine the strength of the relationship between the feature and the target variable. For classification problem, a symmetric AUROC score \(AUC_{sym}\) is defined to evaluate such strength:
ProMS only keeps the features with the top \(\alpha\%\) highest scores. Here \(\alpha\%\) is a hyperparameter that needs to be tuned jointly with other hyperparameters of the final estimator. After the filtering step, data matrix \(D\) is reduced to \(D'\) of size \(n\times p'\) where \(p' \ll p\). To further reduce the redundancy among the remaining features, ProMS groups \(p'\) features into \(k\) clusters with weighted k-medoids clustering in sample space. The \(k\) medoids from each cluster are selected as markers. The whole process is illustrated in the following diagram:
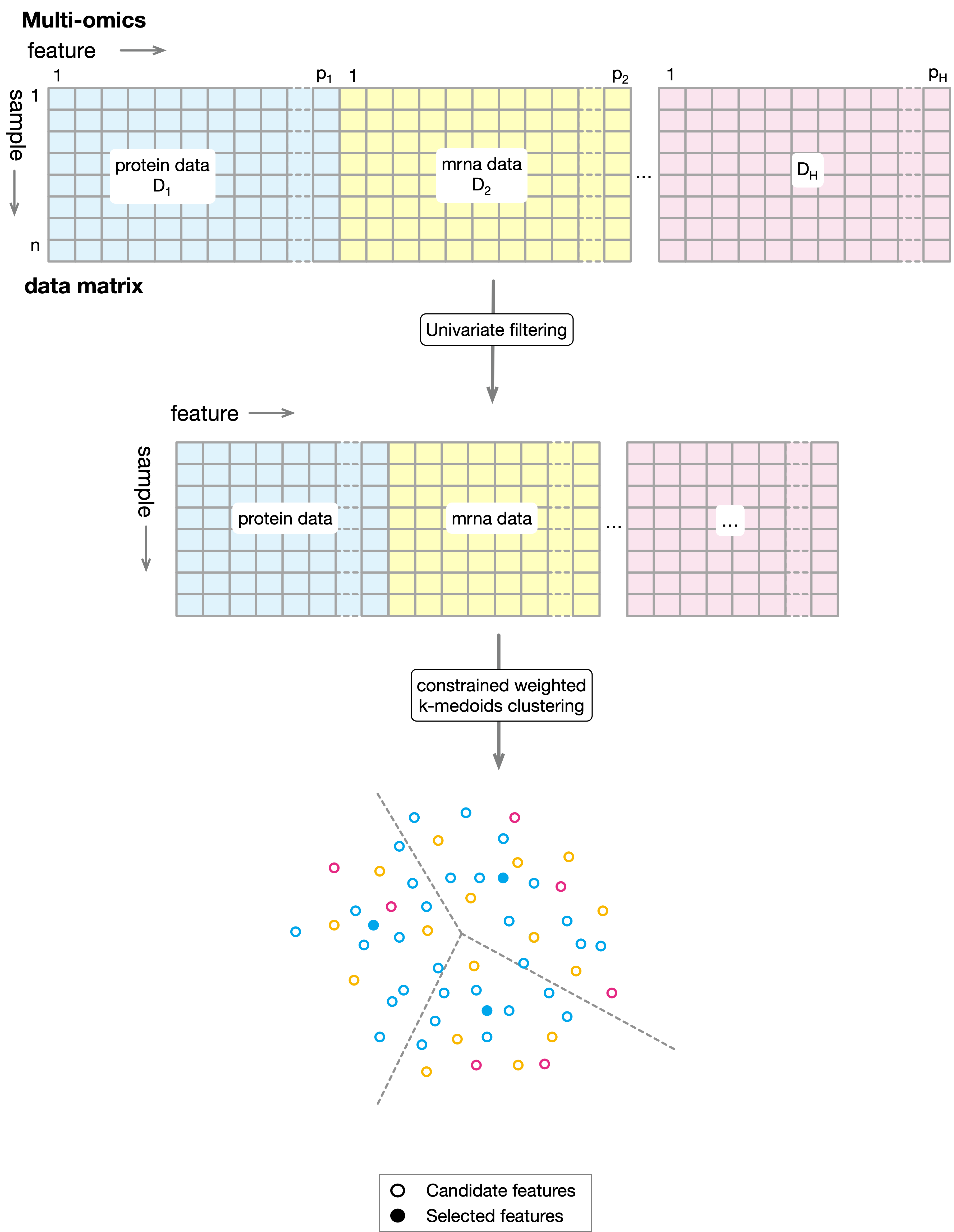
ProMS_mo: Protein marker selection with multi-omics data¶
We have \(H\) data sources, \(D_1, D_2, ..., D_H\) , representing \(H\) different types of omics measurements that jointly depicts the same set of samples \(s_1, s_2, ..., s_n\). \(D_i (i=1,...,H)\) is a matrix of size \(n\times p_i\) where rows correspond to samples and columns correspond to features in \(i\) to represent the proteomics data from which we seek to select a set of informative markers that can be used to predict the target labels. Similar to ProMS, the first step of ProMS_mo involves filtering out insignificant features from each data source separately. Again for classification problem, we use \(AUC_{sym}\) to evaluate feature significance. ProMS_mo first applies the univariate filtering to target data source and keeps only the top \(\alpha\%\) features with the highest scores. We denote the minimal score among these remaining features as \(\theta\). For other data sources, ProMS_mo only keeps those features with score larger than \(\theta\). Filtered data matrices are combined into a new matrix \(D'\) of size \(n\times p'\), where \(p'=\sum_{i=1}^{H} p'_i\) and \(p'_i\) is the number of features in the filtered data source \(i\). Finally, weighted k-medoids clustering is performed to partition the \(p'\) features into \(k\) clusters in sample spaces. To guarantee that only protein markers are selected as medoids, ProMS_mo first selects \(k\) protein markers as the initial medoids. During the iterative steps of optimization, a medoid can only be replaced by another protein marker if such exchange improves the objective function. After the iterative process converges, \(k\) medoids are selected as the final protein markers to train a classifier. The steps are depicted in the following diagram: